
Pairwise Alignment Tutorial
Learn how to align pairs of DNA and protein sequences with Geneious using dotplots and alignment algorithms.
Introduction
The purpose of this module is to develop the skills needed to align pairs of sequences with Geneious. Pairwise sequence alignment allows you to match regions in sequences to identify probable structural and functional similarities.
In this module, we will look at aligning nucleotide (DNA) and polypeptide (protein) sequences using both global (Needleman and Wunsch) and local (Smith and Waterman) alignment methods.
INSTRUCTIONS
To complete the tutorial yourself with included sequence data, download the tutorial and install it by dragging and dropping the zip file into Geneious Prime. Do not unzip the tutorial.
OVERVIEW
Alignment algorithms
EXERCISE 1
Using dotplots to explore relationships
EXERCISE 2
Aligning DNA sequences
EXERCISE 3
Aligning protein sequences

Pairwise Alignment Algorithms
When aligning two sequences, the algorithm will identify the optimal relationship between them. This is done by comparing every letter in one sequence with every letter in the other. The algorithm will account for matches and mismatches and compute the best mathematical path through these matches and mismatches. It will also have to account for insertions or deletions in either sequence. The following two sequences can be aligned by inserting gaps to bring identical residues in line with each other:
Before alignment
AFGIVHKLIVS
AFGIHKIVS
After alignment
AFGIVHKLIVS
AFGI-HK-IVS
The pairwise alignment methods used in Geneious are based on dynamic programming using the Needleman & Wunsch (1970) or Smith & Waterman (1981) algorithms. They are available in global and local variants. A global alignment ensures that every part of two sequences are aligned. A local alignment will align the areas of best similarity such as when only part of the two sequences are related, for example, multi-domain protein sequences. Forcing a global alignment on a multi-domain sequence would not be sensible since the alignment implies that there is a similarity between the sequences for the entire sequence length and parts of the sequences in this case would be unrelated.
Note: An alignment is mathematically optimal and may not necessarily be biologically optimal as you will see in the following exercises.
In addition to the choice of algorithm, you will need to be aware of the scoring scheme and gap penalty settings as these both affect the quality and sensitivity of the alignment method. Scoring matrices allow you to control the sensitivity to mismatches and substitutions during alignment and gap penalties will determine how easy it is for gaps to be opened and extended.
Scoring schemes – For DNA these tend to be quite simple and based on perfect matches and mismatches. Proteins have more complicated substitution tables where similar residue types will score positively even if they do not match exactly, for example, Isoleucine and Leucine. Scoring tables are typically designed to favour a certain level of identity. If your sequences are going to be very similar to each other you can use a strict scoring table but if you want more sensitivity you can use a more relaxed scoring table. With DNA alignments you could choose the scoring scheme that is weighted towards 93% identity where mismatches are scored very negatively and will favour alignments with a high level of identity. Other scoring schemes with lower mismatch scores will be more forgiving of lower identity alignments. Similarly, for protein alignments, you can choose Blosum90 which penalises mismatches and conservative substitutions more than Blosum45 would.
Gap penalties – In addition to the scoring scheme, the gap penalties you select will also affect your alignment quality. The gap open penalty is the cost for starting a gap. Typically this is quite high to discourage gaps. However, the gap extension penalty would be much lower as it would allow gaps to propogate once they have started. This gap open and gap extension scheme is referred to as an affine gap and is intended to clump the gaps together and allow for long runs of gaps. If the gap open penalty is too low then the alignment algorithm will favour opening gaps rather than mismatches and you can end up with a ‘spaghetti’ alignment which is nonsense. Affine gaps are sometimes thought to produce more biologically realistic alignments but you need to balance the cost of opening and extending such gaps and it may be better to use a lower open and higher extend or even have them be equal. Where the two penalties are the same you will be using linear gap penalties where every ‘-‘ costs the same. For certain alignments you might find that it is better to use linear gaps because it allows the program to put single gaps in more cheaply where appropriate (e.g. a beta turn in a protein) when clumping as affines tend to do wouldn’t produce a true alignment.
The balance of gap penalties, scoring schemes and the algorithm you use is the skill you will learn during this module as we explore the effects of these parameters.
Exercise 1: Using dotplots to explore relationships
Dotplots are an excellent visual way to view the regions of similarity between pairs of sequences which cannot be reproduced just by sequence alignment. In particular, identifying repeated regions, inversions and translocations are not handled by pairwise alignment methods.
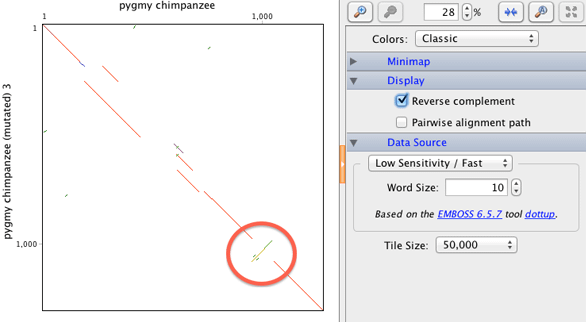
A dotplot marks matches between two sequences on a two dimensional grid. Runs of identity will produce diagonal lines on this grid. To illustrate this, two sequences have been provided. One is an original Pygmy Chimp sequence and the other is the same sequence edited to provide an interesting dotplot.
Select two sequences: pygmy chimpanzee (mutated) 3 and pygmy chimpanzee. You may have to click the tab that says Dotplot. You can also open the dotplot in a new window by clicking the new window button.
What you should see now is a 2D grid with a number of diagonal lines. One sequence appears across the top on the x-axis and the second along the left hand side on the y-axis. The overall trend is for a diagonal from the top left to the bottom right.
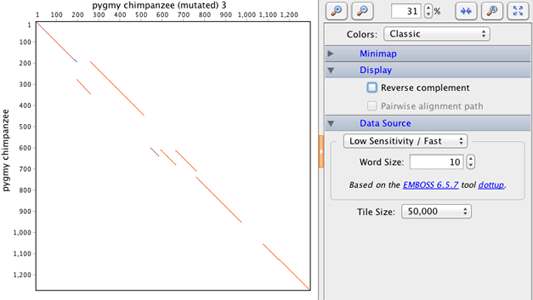
Note: You can zoom in and out on the dotplot by adjusting the zoom level. If you zoom in enough you will be able to see the individual letters of the two sequences on each axis. Also, notice how there is a cross-hair that follows your mouse which gives you the position in the two sequences of your pointer. You can use this to note the location in the dotplot of features you are interested in and this will help you later when you start producing alignments.
Try changing the Data Source settings and see how this affects the dotplot. Notice how changing the sensitivity setting increases or reduces the number of short, noisy points in the dotplot. Increasing the window size will tend to fill the gaps between smaller diagonals to make longer ones. Reducing it will shorten and reduce the diagonals that you see. There may be some interesting detail to be seen but generally reducing this simply adds noise to the plot so increasing the value will leave the strong diagonals visible and get rid of this noise.
Parallel diagonals in a dotplot indicate that there is a repeating pattern in the sequence. A reversed diagonal indicates a sequence inversion event. This means that one sequence has to match against the reverse complement of another sequence.
Now that you are familiar with how these two sequences are related you can move on and start aligning them.
Exercise 2: Aligning DNA sequences
For pairwise alignment, you need two sequences which you expect to have a relationship. In the previous section on dotplots we used two sequences and we will continue with the same ones here. If you haven’t already got the sequences open, select pygmy chimpanzee (mutated) 3 and pygmy chimpanzee.
With the two sequences selected, click the Align/Assemble button on the toolbar. Select Pairwise Align. This will bring up a window that should look like this:
There are a variety of alignment methods available in Geneious. This tutorial uses the Geneious Alignment function. You should reset any changed parameters to their defaults by choosing Reset to Defaults under the Settings cog at the bottom left, unless it is greyed out in which case you already have the defaults selected.
Click OK and the two sequences will be aligned producing a new document. This document will automatically be selected and should look similar to the screenshot below (you may have to turn off the annotations and choose to Wrap the sequences in the sequence viewer options to see the whole alignment more easily).

In addition to the alignment, you also have a dotplot for the two sequences. What you should notice is that the alignment doesn’t look very good in places. There are few gaps and there are regions of high mismatch. This is happening because the default gap penalties are strict and the scoring table is too relaxed. In addition, a pairwise alignment does not deal with sequence inversions.
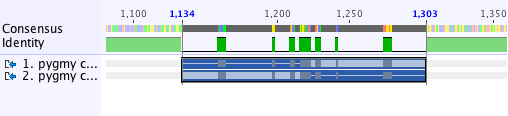
Task 1: In the Alignment View locate the first region of mismatches in your alignment view. Turn on the Consensus sequence and select Highlighting to make this task easier. These options can be found in the Display panel on the right hand side of the Alignment View. In the Highlighting options select Disagreements to Consensus from the dropdown boxes. With these options selected the bases in the alignment which match the consensus sequence will be grey but mismatches will be shaded (mismatches will either appear as black or colored depending on the zoom level on the alignment). Gaps will also be shaded unless you select Ignore gaps in the Consensus options. Mismatching regions will now have a striped effect making them easy to locate. When you have located the first region of mismatches, identify the start and stop coordinates for each sequence and locate that region on the Dotplot.
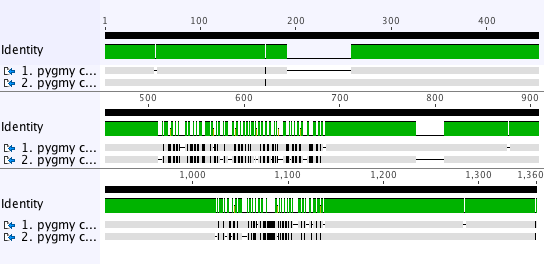
Note how this region of the alignment relates to what you see in that region of the dotplot. The first region of mismatches runs from 443-609 in the original sequence and 511-660 in the mutated sequence. The dotplot shows that this region has some mismatching and then some repeating sequence. Since there are multiple possible paths through this region, the aligner has used the mathematically least costly path but this does not reflect the true relationship since there is clearly a match to be made as indicated in the dotplot.
This task highlights one of the issues with sequence alignments. The alignment will follow the mathematically optimal path through the two sequences and that does not necessarily reflect what has really happened. It is cheaper for the alignment algorithm to force its way through with mismatches than to take the cost of a high gap penalty.
You should realign these sequences with modified settings. Rather than select the original two sequences, you can just select the previous alignment document and select Align/Assemble→Pairwise Align again as a shortcut.
The gap open and extension options are available under the More Options button. The gap extension penalty is set to 3 by default and this will limit the distance that a gap can be extended because every gap extension will subtract something from the overall score. Eventually, a gap will become too expensive compared with just accepting a large number of mismatches and this is exactly what happened with the previous alignment. Another problem is that the default scoring table favors mismatches.
To solve these two problems, change the default cost matrix to 93% similarity and change the gap extension penalty to 0, then click OK.
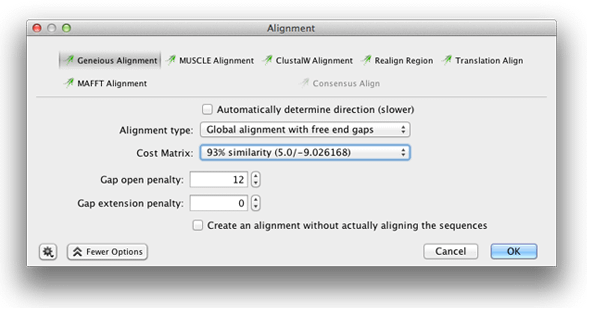
If you look at the alignment now with the same consensus and highlighting options set, you will see that there are almost no disagreement between the two sequences. If you uncheck Ignore gaps it will be clear that the aligner has now used gaps as a way to avoid mismatches.
In the Dotplot there is an inverted region. Locate this region on the new alignment. Remember, a reversed diagonal indicates a sequence inversion event. This means that one sequence has to match against the reverse complement of another sequence. To see the inverted region you will need to select the option Reverse Complement in the dotplot options. Note what the alignment algorithm has done here – in the inverted region, the alignment has simply misaligned the whole section. Pairwise alignments always run in a single direction so for inverted regions you have to be prepared to realign as you will see in the next task.
Task 2: In order to align the inverted region, it is necessary to select just those parts of the sequence. You can do this by clicking on the start of the region and dragging your mouse to the end. Make sure you select the two sequences otherwise you will not be able to extract the alignment region. You selection should look like this:
Click the Extract button and select Extract region alignment. You can then re-align as before but be sure to select Automatically determine sequences direction before clicking OK. Geneious will reverse and complement the second sequence selected before doing the alignment. Compare the dotplot for the extracted region and the re-aligned version. Notice how the diagonal line in the dotplot is now reversed.
Familiarise yourself with the display options that Geneious provides for DNA alignment. Try experimenting with alignment view settings as you will get some interesting effects. For more information on the sequence viewer, look at the relevant Geneious tutorial section.
As we have discovered here, the defaults are not necessarily the best for every situation and those that we have used here are also not ideal in every case. When you are ready, move on to the final exercise.
Exercise 3: Aligning protein sequences
In this exercise you will build on your experience with dotplots and DNA alignments to align a pair of proteins. Select the following two protein sequences: ZP_02833023 and P07990.
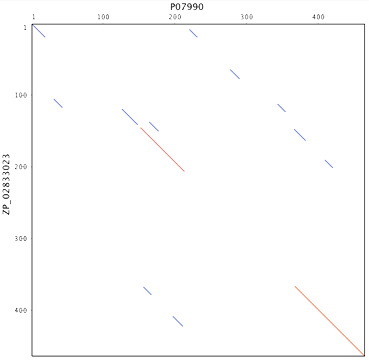
If you are not already in the Dotplot select it now. You may need to adjust sensitivity settings. Select High Sensitivity/Slow and set the window size to 10 and the threshold to 23. This should give you a good idea of where the sequences share common features. You should see two major regions of homology like this (red lines):
Now select Align/Assemble→Pairwise Align to perform a standard protein alignment. Select Geneious Alignment and Reset Defaults if necessary. Click OK and this will create an alignment document. Turn off the annotations and turn on the Identity graph in the Graphs panel. In the Highlighting options select Agreements to Consensus to make the view clearer. You should have a view that looks like this:
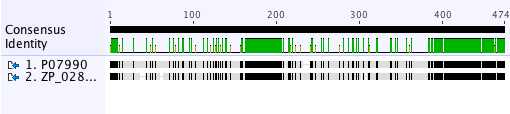
What is the level of pairwise identity between these two sequences and do you think it is a fair representation of their sequence homology?
The pairwise identity can be found in the Statistics panel. The sequences share 43.2% identity. However, there are regions of low identity and other regions of high identity so this number does not truly represent the overall homology.
These sequences are part of two Type I restriction enzymes and are the DNA sequence specificity domains. More information about restriction enzymes can be found here. Examine the alignment and you will see that the level of identity varies greatly along the length of the alignment. The regions that are conserved are the helical spacers shown in this sketch where the specificity subunit is shaded:
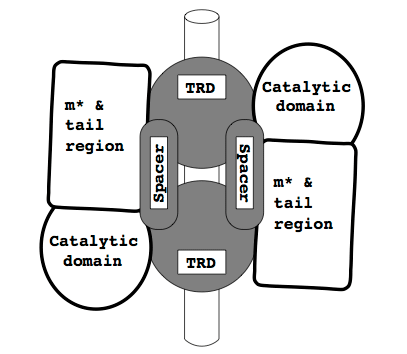
Depending on the family, these helical spacers are highly conserved. However, the Target Recognition Domains are highly variable and this variability is evidence for horizontal transfer. Without the high level of identity in the helical regions it is unlikely that a decent alignment could be made for these two sequences. The TRD regions can be considered to be aligned purely by chance as the identity is so low. The algorithm has simply computed the cheapest path through this region of mismatch.
In this next example, we have two sequences which are distantly related across their entire length. Select P0ACQ4 and P0A2Q4.
Look at the dotplot and you will see that you need to adjust the sensitivity settings significantly to see an obvious diagonal. Hint Try selecting High Sensitivity and increasing the Window Size.
Again, perform an alignment using default settings and examine the result.
Note the % pairwise identity between these two sequences. Is this a fair representation of their homology?
These sequences share 25.2% identity. Mismatches are spread quite evenly throughout the two sequences so this accurately reflects their overall homology.
The identities here are more evenly spread than in the previous example. This alignment is in the twilight zone for a protein alignment although it has features that suggest it is a good match. Clumping of the identities and the fact that the alignment covers most of the length of the two sequences is a good sign. Since both these sequences are annotated, you can turn on annotations and you will see that various regions in the two sequences line up well. Manipulate the view using the zoom functions and other features to observe the details of the alignment and relationship between the sequences.
Look at the pattern of identities and regions of mismatch and note how these relate to the annotated domains and regions. The first part of the alignment has a generally higher level of identity than the second part and this corresponds to the HTH region versus the LysR substrate region. This suggests that conservation needs to be higher in the first domain in order to preserve the function.
There is a region labelled “HTH 1 Region” in both sequences. Examine how the pattern of matches and mismatches relate to periodicity in alpha helical structures. Turn on the hydrophobicity graph and see if there us a relationship between this and the pattern of matches. The HTH 1 Region is helical and there are 3.6 residues per turn of a helix. This periodicity does appear to be reflected in the conservation of matches. The hydrophobicity graph supports this as the hydrophobic residues tend to be conserved so as the backbone moves from buried to exposed in the helix the buried residues are conserved and the exposed residues are variable.
One of these sequences has secondary structure annotations for the LysR Substrate region. Looking at this alignment and other evidence such as patterns of matches, would you think it is likely that the two sequences share the detailed secondary structure? The annotated domains line up nicely and the pattern of hydrophicity supports the pattern of secondary structure elements so it is likely that the secondary structure and tertiary structure of the two proteins is very similar despite the low overall identity.
Realign these two sequences using a strict Blosum90 table and Smith Waterman algorithm. Look at what has happened to the alignment identity and length. Aligning these two sequences with Blosum90 and Smith and Waterman results in the alignment being truncated and the reported sequence identity has increased. This is because the Blosum90 scoring table penalises mismatches more than Blosum62. For a distantly related pair of sequences, this will tend to result in a lower overall score and thus regions of poor match become harder to traverse. The local similarity algorithm combined with this lower scoring prevents the alignment of the lower identity C-terminal ends of the two sequences resulting in the higher apparent identity (28.1%). Although the dotplot shows that there is a match beyond approximately residue 190 in each sequence, this stricter alignment is not able to bridge the gap because the score benefit is insufficient.
You should realise now that alignments are not a simple process and that alignments do not necessarily represent biological truth. Use them in conjunction with other available information and take care especially when working with distantly related sequences. This will help you understand what is going on with the search parameters and alignments in BLAST searches and also when you start working with multiple alignments.
This concludes the pairwise alignment tutorial.