
Molecular Analysis of Plant DNA Tutorial
Learn how to annotate plant DNA sequences using BLAST and multiple alignment, then read a phylogenetic tree to explore genetic distance.
Written by Dr Jack da Silva, University of Adelaide, Australia.
Introduction
In this tutorial, you will use Geneious Prime to analyze plant DNA sequences by database searching with BLAST, building a multiple alignment and constructing an evolutionary tree.
The plant sequences provided in this tutorial are from the Fabaceae family of flowering plants. These plants are legumes and include many important food crops such as alfalfa (Medico sativa), and clover (Trifolium spp).
INSTRUCTIONS
To complete the tutorial yourself with included sequence data, download the tutorial and install it by dragging and dropping the zip file into Geneious Prime. Do not unzip the tutorial.
EXERCISE 1
Database Searching with BLAST
EXERCISE 2
Multiple Sequence Alignment
EXERCISE 3
Molecular Phylogenetics

Database Searching with BLAST
In this section you will perform a database search using one of the provided plant DNA sequences.
BLAST stands for Basic Local Alignment Search Tool. It is a computer method that takes a sequence provided by the user, called the query sequence, and searches a database of sequences to find close matches. The databases most often used are the public nucleotide sequence repositories located in North America (NCBI GenBank), Europe (EMBL), and Japan (DDBJ).
BLAST searches are conducted for a variety of reasons. You may want to find similar sequences in order to analyse their evolutionary history. Or, as in our case, you may want to find similar sequences whose structures and functions have been deciphered in order to make inferences about your query sequence.
You can run a Blast search against NCBI GenBank from within Geneious.
Open one of the provided plant DNA sequences. Notice the sequence document provides nothing beyond the name and sequence and that there are no annotations on this sequence. By running a BLAST search with this sequence, we hope to find out what is known about this sequence or a close relative.
You can BLAST this sequence by clicking BLAST in the top tool bar. You should reset any changed parameters to their defaults by choosing Reset to Defaults under the settings button at the bottom left of the BLAST window – unless it is greyed out in which case you already have the defaults selected. We will run a BLAST search against the Nucleotide collection (nr/nt) database using the blastn program. The “nr” database refers to the “non-redundant” part of GenBank although this is historical only because the database is no longer non-redundant and contains all of the sequences found in GenBank+RefSeq Nucleotides+EMBL+DDBJ+PDB but no EST, STS, GSS or phase 0,1 or 2 HTGS sequences.
Keep all other settings in the BLAST options at their default values and click the search button. The search may take a few minutes to run.
Examine your search results
Once the search finishes, Geneious will download the alignments into a results folder, which will be a subfolder of the folder containing your query. The results can be sorted in a variety of ways. Sort them by “E Value” – this should be the first column. The sorting should be set so that the least likely hit is at the top (ie. the small triangular arrow will point up).
Look at what happens to the Bit-Score, % Pairwise Identity and so on as you scroll down the list (you may need to scroll across to see these values).
Above all, you should always look at the alignments. Click through some of the hits to view the alignments. The Alignment View shows the query sequence aligned to the database (result) sequence (for more information on pairwise alignments, see the Pairwise Alignments tutorial). You should notice here that the length of the alignment differs from the full length of the database sequence for many of the hits. To see the base numbers for the hit and the query, open the Advanced settings tab (on the right hand side of the alignment view window). Tick the box next to Numbering and in the dropdown box next to this select All Sequences. The alignment length differs because BLAST implements a local alignment which means it does not try to align whole sequences, just the parts that match best. For BLAST hits to be truly signficant, the alignment should be a substantial subset of the original aligned pairs and there should be good levels of identity.
The Alignment View shows a graph displaying the identity between the query sequence and the hit. To view the identity graph, open the graphs tab (on the right hand side of the alignment view window) and tick the boxes next to Show graphs and Identity.
If you wish to display all of the BLAST hits against the query sequence in one view, click the Query-centric view tab at the top of the Document table. In this view the query sequence is displayed as a reference sequence with all the hits mapped to it.
Switch back to the Hit Table view, then select the hit to Medicago polymorpha, Accession number DQ311981 (you can filter the hit table using the Search function in the top right hand corner of Geneious). Click the Download full sequences button in the sequence viewer. Geneious will download the full NCBI GenBank record for this sequence including available annotations and add a Sequence View tab which shows the hit sequence annotated with “BLAST hit” where the query matches. If the annotations are not currently visible, open the annotations tab and check that the boxes next to Show annotations and next to each of the annotation types are ticked.
Click on the Text View tab to see the NCBI record (you may have to scroll down past the alignment). This text record provides the information for Geneious to display its graphical view, in particular the annotations shown against the sequence in the “Sequence View” tab.
Copy this sequence to your plant DNA folder by simply dragging the sequence entry in the Document Table to your folder.
Multiple Sequence Alignment
Before sequences can be compared, they must be aligned. This is a trivial exercise if two sequences are from exactly the same genome region, are of the same length and neither has had any nucleotide insertions or deletions. In this case, simply lining up the two sequences automatically aligns each identical nucleotide site.
Unfortunately, the simple situation described above is rarely found, as you have observed when comparing your query sequence to database sequences in the results of your BLAST search. Misalignment between sequences may occur for several reasons. The sequences may be from slightly different, but overlapping, genome regions. Or, more commonly, the sequences are from the same genome region, but, as a result of mutations, one or both sequences have experienced one or more nucleotide insertions or deletions.
The objective of sequence alignment is to align homologous residues among two or more sequences. This is accomplished by placing gaps in one or both sequences so as to increase their similarity at each site, and to do so using the fewest number of gaps. A gap at a nucleotide site in one sequence represents either a deletion in that sequence or an insertion in the other sequence. Since it is usually unknown which event occurred, events resulting in caps are referred to as indels (insertion or deletion). Because indels are rare relative to nucleotide changes, gaps are inserted into sequences sparingly. This is accomplished by using a method that assigns “costs” to initiate and extend a gap and then charges these costs against the increased similarity of two sequences.
Build the alignment
You will now use Geneious Prime to align the plant DNA sequences provided plus the new sequence you found in the BLAST search section.
The tutorial folder contains a set of 22 DNA sequences which can be identified by the nucleotide icon. In addition, the sequence from your BLAST search will also be a DNA sequence with the same icon. Select all of the DNA sequences in the tutorial folder. Make sure you do not select the tutorial document itself because if you do, the alignment button will not work.
With all 23 sequences selected, click on Align/Assemble→Multiple Align (or in the Tools menu if you prefer). We will perform an alignment using MUSCLE as it is faster than the Geneious aligner, so select this option. Keep all the other settings at their defaults.
Click the OK button. After the alignment is completed, the alignment document, will appear in the Document Viewer panel and the document should open automatically.
Inspect the alignment in the Alignment View tab.
View the annotations of the sequence you imported from the BLAST search in the alignment. If you cannot see the annotations you need to turn on annotations in the Annotations and Tracks panel to the left of the alignment viewer.
Expand the % Statistics panel on the right side of the Alignment View tab.
Molecular Phylogenetics – Construct an evolutionary tree
Multiple sequence alignment is most often used as a preliminary step in determining the evolutionary relationships among the sequences in the alignment. The study of these relationships among molecular sequences is called molecular phylogenetics. The objective of molecular phylogenetics is to infer the evolutionary history of a given set of sequences. Evolutionary history is usually represented by a diagram of hierarchically nested branches that depicts the series of events (e.g. speciation or mutation) that lead to the sequences under study. Such a diagram is called an evolutionary tree or a phylogeny.
Select the multiple alignment you constructed in the previous exercise and click the Tree button in the tool bar. This opens the Tree Dialogue box as shown here:
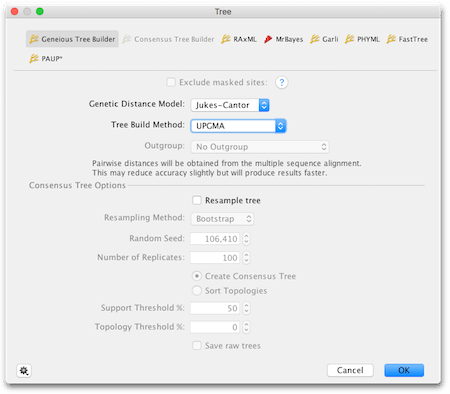
Before changing anything, click “Reset to defaults” under the settings cog in the bottom left corner of the box.
Geneious has a variety of tree building methods available, some of which can be installed as plugins. We are going to use the Geneious Tree Builder which builds distance based trees. We will be using distance methods because they are intuitively simple and fast and are useful for a preliminary analysis.
For the Genetic Distance Model option, we will use Jukes-Cantor. This is the simplest model and assumes equal rates of change between all nucleotides.
For the Tree Build Method option, choose UPGMA.
For more information on these options see the Geneious phylogenetics tutorial.
Leave all other options with their default values and then click OK
Geneious will build the tree and create a tree document. Click on this document to open it, if it has not already automatically opened in Geneious.
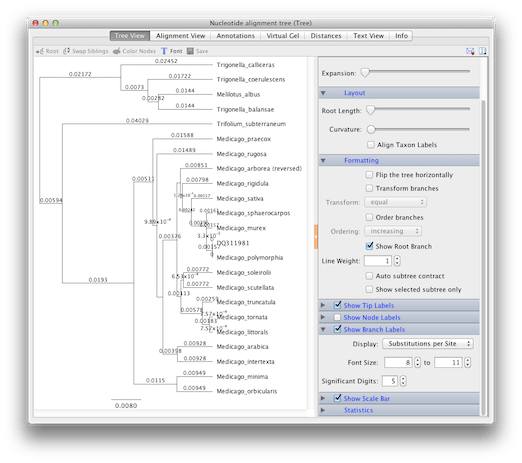
In this view, evolution proceeds from left to right, with all of the sequences having a common ancestor.
Note that the vertical lines in this diagram have no meaning – they are used simply to separate the horizontal lines and sequence names.
The horizontal lines are branches and their lengths are scaled to the amount of evolutionary change. In this case, evolutionary change means the substitution of one nucleotide by another at a particular site and is measured as the number of substitutions per site. Note the scale bar at the bottom of the diagram (if you can’t see it, select Show Scale Bar in the right-hand panel).
You’ll notice the sequence that you downloaded from Genbank (DQ311981) is only separated from Medicago polymorphia by a vertical branch. This is because the GenBank sequence is from the same species so the branch length between these sequences is zero
To see individual branch lengths, select Show Branch Labels in the right-hand panel. Note that branch lengths of these trees are given in scientific notation. For example, 2.13 x 10-2 = 0.0213.
The way to interpret this diagram is that two sequences clustered together share a more recent common ancestor than either does with any sequence outside the cluster.
View the Distance matrix for the tree by clicking the Distances tab at the top of the Document Viewer panel. The Distance Matrix shows the genetic distance between each pair of sequences. These are the distances that were used to construct the tree.